

Enfermedad de Gaucher: Complicaciones
Complicaciones en la Enfermedad de Gaucher
Introducción:
La enfermedad de Gaucher (EG) es una enfermedad de almacenamiento lisosómico autosómica recesiva, causada por la deficiencia de la enzima glucocerebrosidasa necesaria para la degradación de los glucoesfingolípidos.
Las manifestaciónes clinicas incluyen hepato-esplenomegalia trombocitopenia, enfermedad ósea y diátesis hemorrágica, que con frecuencia resultan en consulta con los hematólogos.
El desarrollo de la TRE intravenosa en la década de 1990 ha dado como resultado mejoras dramáticas en las complicaciones hematológicas y vicserales. el reconocimiento de complicaciones, inlcuido el mieloma múltiple y la enfermedad de Parkinson, ha desafiado la vision tradicional en los macrófagos de la fisiopatología de este trastorno.
Complicaciones hematológicas:
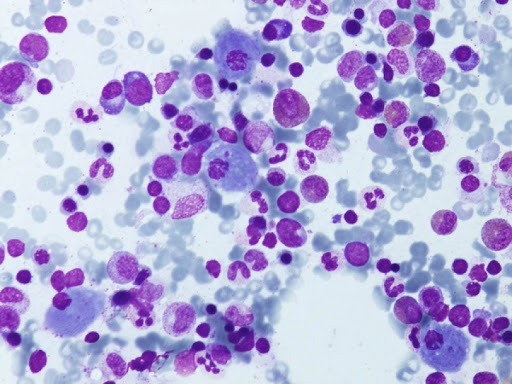
Un número significativo de pacientes con EG atendidos por hematólogos son detectados por medio de biopsia de médula ósea, que revela la presencia de macrófagos característicos congestionados de lípidos que expresan fosfatasa ácida, CD68, CD14 y HLA clase II, pero no CD11b, CD40 o marcadores de células dendríticas. La anemia, la trombocitopenia y, en menor medida, la leucopenia son una consecuencia de la infiltración médula ósea, agravada por el secuestro esplénico, y pueden observarse de forma siultánea o independiente. Como ocurre con la anemia, la trombocitopenia también está relaciomada con el hiperesperismo y/o la infiltración de la médula ósea, pero sin correlación directa con el grado de esplenomegalia.
También se han descrito pacientes con EG y anemia hemolítica autoinmune o púrpura trompocitopénica idiopática y, por lo tanto, se deben excluir las complicaciónes inmunitarias en casos atípicos en los que el curso clínico se asocia con una disminución rápida de los recuentos sanguineos o una falla en la mejora con la terapia de Gaucher.
El tratamiento de la anemia relacionada con la EG ha dependido históricamente de transfusiones de hemoderivados, esplenectomía y, más recientemente, terapia de TRE para restaurar la hemoglobina a los objetivos terapéuticos acordados.
Los pacientes con EG tienen riesgo de hemorragia, causada principalmente por trombocitopenia grave; sin embargo, se ha observado que la extensión de la tendencia al sangrado es desproporcionada con el recuento de plaquetas, lo que sugiere anomalías concurrentes de la función plaquetaria y factores de coagulación.
También se ha encontrado que los pacientes con EG de tipo 1 tienen anomalías en la función plaquetarias incluida una adhesión plaquetaria significativamente menor, asociada con hemorragia mucosa; pruebas de mezcla mostraron que la adhesión reducida era un defecto plaquetario
intrínseco que no fue afectado por el uso de TRE. Las anomalías de los factores hemostáticos, el número reducido de plaquetas y la disfunción dan como resultado un mayor riesgo de hemorragia, por ejemplo, durante procedimientos dentales y de cirugía
mayor.
La atención a la hemotasia local ha permitido procedimientos invasivos seguros, con transfusiones de plaquetas administradas solo a pacientes considerados de alto riesgo sobre la base de una puntuación de hemorragia.
El embarazo también tiene potencial de exacerbar las manifestaciones de la EG y el riesgo de hemorragia durante el parto puede aumentar en las mujeres con esta enfermedad.
El bazo en la EG:
La esplenomegalia está presente en aproximadamente el 90% de los pacientes y puede ser masiva, con un volumen esplénico medio de 15,2 veces lo normal.
La esplenomegalia sintomática con dolor, la distensión abdominal o citopenias debido a la acumulación esplénica pueden ser las características de presentacíón de la EG.
Las lesiónes esplénicas focales se encuentran en 20 a 33% y su incidencia aumentada con la edad. En la mayoria de los pacientes, estas lesiones, que probablemente representan una fibrosis focal, persistirán a pesar de la TRE.
La TRE tiene como objetivo eliminar la esplenomegalia y el hiperesplenismo sintomático con una disminución del 50% en el volumen del bazo alcanzable dentro de los dos años de terapia.
El agrandamiento del hígado y el bazo es común en la enfermedad de Gaucher tipo 1 no tratada, y se pueden observar nódulos esplénicos que pueden confundirse con malignidad, particularmente en individuos con esplenomegalia masiva (fig 1). La función hepática suele ser normal, aunque se puede observar hiperbilirrubinemia en pacientes de ascendencia judía Askenazi debido al sindrome de Gilbert.
Esplenectomía en EG:
Antes de la llegada de la TRE, se realizaba una esplenectomía terapéutica para la esplenomegalia sintomática y el hiperesplenismo severo.
En la actualidad la esplenectomía rara vez se realiza y no debe realizarse a la ligera, ya que puede provocar un empeoramiento sustancial de la enfermedad en otros órganos.
El deterioro de la enfermedad ósea a menudo ocurre después de la esplenectomía y los pacientes con EG esplenectomizados también tienen un mayor riesgo de Hhipertensión pulmonar.
Dentro de la población con EG, la esplenectomía se ha asociado con un mayor riesgo de malignidad, que probablemente se relacione con la propia EG ya que la esplenectomía desúés de un traumatismo en individuos sanos no se ha asociado con un aumento de la malignidad.
Gammapatías y malignidad:
Existe una mayor incidencia de gammapatías policlonales y monoclonales en la EG con un mayor riesgo de Mieloma Múltiple, La gammaglobulinemia policlonal es común en el momento del diagnóstico, reportada de 14 a 41% de los adutos, siendo las anomalías de IgG las mas comunes.
Se ha informado una mayor prevalencia de gammapatía monoclonal de significado indeterminado (GMSI), de hasta un 25%, en pacientes con EG y al igual que en la población general, es más común en pacientes de edad avanzada.
Se informó una alta incidencia de reordenamientos en el locus del gen de la cadena pesada de inmunoglobulina (IGH) (41%) en una cohorte de pacientes con EG con una edad media de 34 años, de los de los cuales no tenían valor de paraproteína medible, lo que sugiere que la expansión clonal puede ocurrir relativamente temprano en el curso de la enfermedad.
Se ha observado que el riesgo aumentado de mieloma múltiple varia de 5,9 veces a 51,1 veces más que el de la población normal. Las asociaciones entre la EG y la neoplasias malignas de órganos sólidos son menos comunes, pero varios estudios han informado una mayor incidencia de carcinoma hepatocellular.
La terapia de reemplazo enzimático da como resultado una mejora significativa en las gammapatías policlonales tanto en adultos como en niños, pero las paraproteínas generalmente permanecen detectables. Si bien existe una superposición entre algunas características de malignidad y EG, como dolor óseo, esplenomegalia y citopenias, se requiere un seguimiento a más largo plazo para determinar cualquier efecto de la TRE sobre el riesgo de malignidad.
Complicaciones óseas:
La afectación ósea es una fuente de morbilidad significativa entre los pacientes con enfermedad progresiva no tratados (Fig 2). Las complicaiones reportadas incluyen osteoporosis, fractura por compresión vertebral y osteonecrosis de la cabeza femoral. Clínicamente, los pacientes pueden experimentar un episodio de dolor óseo que afecte a una extremidad o articulación, similar al observado en pacientes con anemia de células Falciformes. Los hallazgos locales, como eritema e hinchazón, requieren una distinción de la osteomielitis.
Un trabajo reciente ha definido más claramente los factores de riesgo de enfermedad ósea significativa, incluída la esplenectomía, y confirmó de manera importante la relación entre la baja densidad ósea y el riesgo de fractura en la EG.
Conclusiones:
La EG es un trastorno poco común, pero tiene el potencial de influir en la comprensión de los trastornos más comunes, incluidas ciertas neoplasias malignas y la enfermedad de Parkinson.
Una vez conocido predominantemente por su aparición como causa de esplenomegalia o por su aspecto característico de la molécula ósea en los exámenes médicos, el éxito de TRE y los descubrimientos científicos que sustentaron ese éxito marcaron el comienzo de una nueva era en la terapéutica celular. TRE para GD se ha considerado un prototipo en la búsqueda de desarrollar terapias similares para otras deficiencias enzimáticas hereditarias.
Las estrategias de tratamiento más nuevas en desarrollo, que incluyen terapia de reducción de sustrato (TRS), acompañantes y terapia génica, pueden ofrecer nuevas opciones terapéuticas a los pacientes con EG y sus medicos tienen el potencial de una transferibilidad aún más amplia a otros trastornos con una base genética o metabólica.
El creciente reconocimiento de otras complicaciones de la EG, en particular el mieloma múltiple y la enfermedad de Parkinson, sugiere que el metabolismo aberrante de los glucoesfingolípidos puede desempeñar un papel en la patogenia de trastornos más comunes.
Las vias por las que la deficiencia enzimática da lugar a las manifestaciónes clínicas de este trastorno no son del todo conocidas, sin embargo se ha relacionado con los perfiles alterados de citocinas inflamatorias, derivados de esfingolípidos biocativos y alteraciones en el microambiente de la médula ósea.
El esclarecimiento adicional de estas vías contribuirá para mejorar nuestra compresión no solo de la EG, sino también de los trastornos asociados.


Lo sentimos, el formulario de comentarios está cerrado en este momento.