

Enfermedad de Gaucher: Diagnóstico
Establecer el diagnóstico inicial de la EG puede ser un desafío debido a las presentaciones fenotípicas altamente variables y la superposición con otros trastornos. Esto se complica aún más por un número creciente de niños asintomáticos que son diagnosticados mediante exámenes genéticos. Dado que el tratamiento para la EG puede ser costoso, lento e invasivo, decidir cuándo y cómo comenzar el tratamiento puede ser difícil. Una comprensión más clara de los matices de las diferentes presentaciones de la enfermedad a diferentes edades puede conducir a un mejor tratamiento y asesoramiento al paciente / padre.
Diagnóstico pediátrico a diferentes edades de la EG
Cribado prenatal:
La detección prenatal de los trastornos por almacenamiento lisosómico (TAL) se realiza con mayor frecuencia en poblaciones específicas como por ejemplo la población Judía Asquenazí, en las cuales se recomienda realizar un cribado de 4 a 8 mutaciones comunes del gen que codifica para la enzima lisosomal glucocerebrosidasa 1 (GBA1), en donde se pueden identificar hasta el 95% de los alelos mutantes, lo que hace que dichos cribados sean bastante eficaces. En poblaciones con diferentes orígenes étnicos, estos paneles identifican un porcentajemucho menor de mutaciones causantes de enfermedades. Por lo tanto, se recomienda la secuenciación de todo el gen GBA1. En las familias afectadas, donde se conocen las mutaciones de GBA1, el cribado prenatal se puede realizar fácilmente para detectar mutaciones especificas en los portadores.
Los embarazos posteriores pueden examinarse mediante muestras de vellosidades coriónicas o líquido amniótico. Si no se conocen las mutaciones, el diagnóstico prenatal se puede realizar mediante ensayos enzimáticos en un laboratorio experimentado.1
Cribado en recién nacidos:
La detección de trastornos como la EG en recién nacidos es un tema muy debatido, ya que dichas pruebas también identificarán a los bebés que pueden estar asintomáticos durante años o incluso toda la vida. La detección temprana de este trastorno puede generar beneficios debido al uso de tratamiento tempranos para controlar los síntomas y así prevenir numerosas secuelas a largo plazo.1
Presentaciones sintomáticas durante el primer año de vida:
Tanto la EG neuronopática aguda como la crónica, así como la no neuronopática, pueden manifestarse en la infancia media o tardía. En la EG2 clásica, los lactantes pueden parecer normales durante el período neonatal inmediato, pero comienzan a desarrollar los síntomas descritos en la sección anterior sobre el inicio neonatal, y la muerte generalmente se produce en los primeros 18 meses. Se pueden utilizar tratamientos más agresivos como la traqueotomía, gastrostomía y terapia de reemplazo enzimático (TRE), lo cual ha aumentado el tiempo y la calidad de vida. Sin embargo, suele seguir la detención o la regresión de los hitos del desarrollo, las convulsiones y las mioclonías. Si bien los síntomas viscerales se pueden mejorar con la TRE, hay poco impacto en la neurodegeneración, ya que la TRE no atraviesa la barrera hematoencefálica (BHE). Los pacientes con EG3 a menudo se presentan durante el primer año de vida con organomegalia masiva, anemia o trombocitopenia y una parálisis de la mirada supranuclear horizontal. La mayoría de los niños alcanzan los hitos del desarrollo, aunque su capacidad para gatear o caminar puede verse obstaculizada por su organomegalia masiva. Estos niños, en contraste con aquellos con EG2, muestran una mejora notable con el inicio temprano de la TER. Con frecuencia se observa reversión significativa de las manifestaciones no neurológicas, una rápida recuperación de las habilidades motoras y un aumento de peso. En pacientes con EG3 con neurodegeneración, la progresión, aunque no se ve afectada por ningún tratamiento disponible, es marcadamente más lenta que en EG2. Los pacientes con EG1 también pueden presentarse en el primer año de vida. Por lo general, primero se observa que tienen organomegalia o citopenia, a menudo después de una enfermedad viral no relacionada. En los últimos años, ha sido posible establecer el diagnóstico de EG mediante análisis de gotas de sangre seca. Esto ha permitido a los médicos diagnosticar a pacientes que no tienen acceso a las instalaciones de pruebas adecuadas en regiones geográficamente remotas.
Estas gotas de sangre seca también se pueden usar para medir los niveles de glucosilsfingosina, que se muestran prometedores como biomarcador de la gravedad de la enfermedad y la respuesta al tratamiento.1
Presentaciones sintomáticas en la infancia y la adolescencia.
Tanto la EG1 como la EG3 pueden presentarse en la edad pediátrica, aunque la EG1 es más común. En la infancia y la adolescencia, los síntomas de presentación de EG1 son principalmente hematológicos, lo que conduce a un diagnóstico erróneo frecuente de EG1 como un trastorno hematológico / oncológico. Estos síntomas pueden incluir anemia, trombocitopenia, hemorragias nasales, sangrado excesivo y esplenomegalia. La esplenomegalia está presente en aproximadamente el 95% de los niños diagnosticados de EG y suele aparecer antes de otras manifestaciones. La EG3 puede presentarse en la infancia con esplenomegalia, anemia, trombocitopenia, crisis óseas o cifosis, junto con manifestaciones neurológicas.
En un subconjunto raro de pacientes con EG3, puede desarrollarse una afectación grave de la válvula cardíaca. El diagnóstico de EG3 lo realiza con frecuencia un neurooftalmólogo cuando se observan movimientos oculares anormales. Se recomienda que el tratamiento comience temprano en niños sintomáticos con EG1 y EG3 para evitar daño óseo y visceral irreversible, así como otros problemas de crecimiento y desarrollo a largo plazo. La baja estatura o el retraso del crecimiento son frecuentes en pacientes con EG1 y EG3. Las manifestaciones óseas en la EG, que suelen aparecer en la adolescencia, son variadas y pueden incluir osteonecrosis, crisis de dolor óseo, lesiones líticas, osteoporosis (a lo largo de la vida), fracturas patológicas y deformidad en matraz de Erlenmeyer. Se estima que hasta el 75- 90% de los pacientes diagnosticados con EG experimentarán algunos hallazgos óseos durante el curso de su enfermedad. El tratamiento durante la adolescencia, cuando sea necesario, puede mejorar algunos de estos efectos, aumentando la salud y la estabilidad de los huesos más adelante en la vida.
Diagnóstico y presentaciones sintomáticas en la edad adulta:
Los síntomas que se presentan en la edad adulta suelen ser similares a los de la infancia e incluyen anemia, trombocitopenia, hepatoesplenomegalia y manifestaciones óseas. En general, se considera que los pacientes que solo desarrollan síntomas en la edad adulta tienen una EG más leve, pero hay excepciones.
Pruebas para diagnosticar la EG
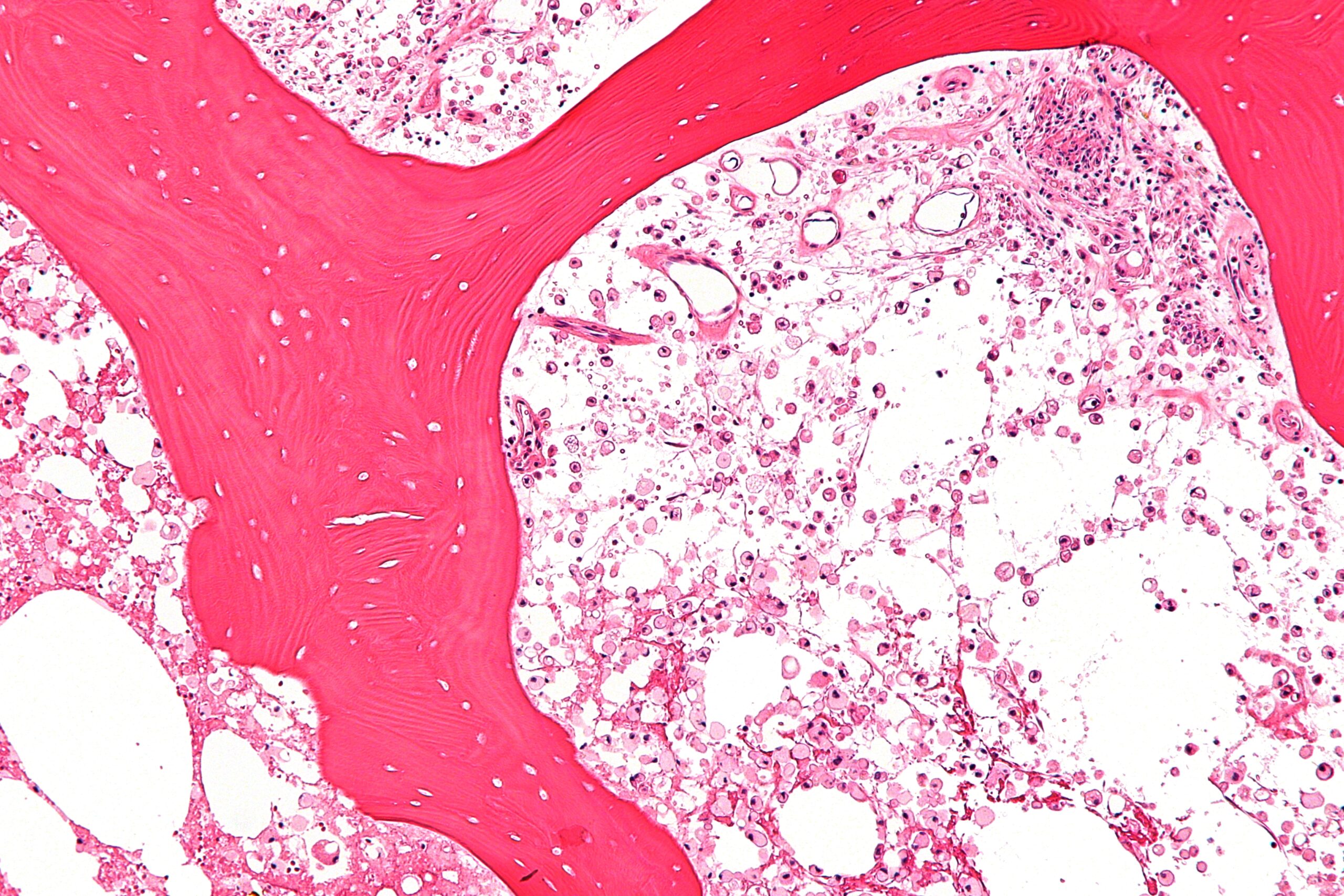
Biopsia de médula ósea: históricamente, los pacientes con EG se diagnosticaban mediante un examen de la médula ósea, ya que la combinación de anemia, trombocitopenia y / o esplenomegalia sugería una neoplasia maligna hematológica. Las células de Gaucher pueden verse en una biopsia de médula ósea de pacientes con EG y se tiñen positivamente con reactivo de ácido periódico-Schiff (PAS). Sin embargo, la biopsia de médula ósea ahora solo está indicada cuando se sospecha una comorbilidad hematológica.3 Actividad de la enzima ?-glucosilceramidasa: la actividad enzimática, el estándar de oro para el diagnóstico de EG, generalmente se mide en leucocitos de sangre periférica utilizando el sustrato 4-metilumbeliferil-?-D-glucopiranósido en un ensayo fluorométrico. En pacientes con EG, la actividad de la enzima glucosilceramidasa en los leucocitos de sangre periférica es del 0% al 15% de la actividad normal. No existe correlación entre el nivel de actividad enzimática y la gravedad o el tipo de EG. Los portadores tienen una actividad enzimática intermedia entre los niveles normales y afectados de EG. La actividad enzimática es una prueba poco fiable para la detección de portadores debido a la superposición de los niveles de actividad enzimática entre portadores y no portadores y entre portadores e individuos afectados.
Pruebas de genética molecular: la deficiencia de la glucocerebrosidasa en la EG surge de mutaciones bialélicas en el gen GBA, excepto en raras personas con deficiencia de sapocina C. En el 99% de los pacientes con EG, la secuenciación completa del gen de la GBA revelará sus dos mutaciones causales. Se sabe que más de 300 mutaciones causan EG. Cuando se sospecha de EG, se debe medir la actividad de la enzima ?-glucosidasa ácida leucocitaria en sangre junto con las pruebas genéticas moleculares que brindan información de pronóstico y facilitan la detección familiar.3 Correlación genotipo-fenotipo: la correlación genotipo-fenotipo en la EG no es absoluta, pero puede ayudar en el asesoramiento y el tratamiento de los pacientes con EG. N370S es la mutación GBA más común y más leve. La presencia de al menos una mutación N370S predice con precisión EG1, mientras que la homocigosidad para la mutación L444P predice con precisión la EG neuronopática, aunque no es posible establecer si el paciente tiene EG2 o EG3. La mutación N370S representa aproximadamente 80 % de alelos de la enfermedad en un estado homocigoto (N370S / N370S) o en un estado heterocigótico. Los pacientes que son homocigotos para N370S suelen tener una enfermedad más leve; sin embargo, algunos pacientes pueden presentar síntomas graves. Los pacientes con afectación cardiovascular son invariablemente homocigotos para la mutación D409H. Se presume que la homocigosidad para las mutaciones 84GG o IVS2 + 1 es letal. Las mutaciones 84GG y V394L son exclusivas de los pacientes que provienen de la población Judía Ashkenazi.3 Monitoreo de enfermedades: la evaluación de la gravedad general de la EG requiere una evaluación completa de todos los sistemas de órganos involucrados en el proceso de la enfermedad. Se debe considerar la importancia de las clínicas designadas para la EG en las que los médicos que siguen a estos pacientes conocen la enfermedad y las opciones de tratamiento. En lugares donde la enfermedad es rara y las distancias son grandes, el paciente debe ser monitoreadode cerca por un médico local y tener una consulta con un experto en EG una vez al año o si se desarrollan complicaciones. Se debe realizar exploración física y revisión de la historia clínica cada 6-12 meses con especial atención a episodios hemorrágicos, dolor óseo, hepatoesplenomegalia, crecimiento en niños y evaluación neurológica especialmente en los tipos neurológicos de EG. Es necesario un recuento sanguíneo completo para buscar anemia y trombocitopenia. Se recomienda la evaluación del volumen del hígado y el bazo mediante resonancia magnética (RM). La ecografía abdominal también se puede usar cuando la resonancia magnética no está disponible, pero es una herramienta inadecuada para medir el volumen. Clásicamente, las radiografías simples del fémur distal pueden demostrar la deformidad en matraz de Erlenmeyer. El seguimiento del esqueleto con resonancia magnética de la columna y el fémur revela la extensión de la afectación de la médula ósea y la presencia de fibrosis o infarto. La Puntuación de carga de médula ósea se puede utilizar para realizar un seguimiento de la afectación de la médula ósea del paciente mediante RM. La absorciometría de rayos X de energía dual (DEXA) de la columna lumbar y las caderas se utiliza para evaluar la densidad mineral ósea.
Biomarcadores séricos en la EG:
Las medidas objetivas que reflejan la gravedad de la enfermedad y puedan evaluarse como resultados terapéuticos son necesarias para evaluar la EG y desarrollar una estrategia de tratamiento. Esto puede ser cada vez más importante a medida que se diagnostican más pacientes presintomáticamente a través de diferentes programas de detección. Los biomarcadores evaluados cada 6-12 meses pueden evaluar la progresión clínica y la estabilización de la enfermedad del paciente. La quitotriosidasa se ha utilizado como biomarcador durante más de una década, pero algunos pacientes tienen una deficiencia hereditaria de esta enzima. Actualmente, LysoGB1 parece ser el biomarcador más sensible y predictivo de los síntomas de la EG, como la trombocitopenia y la esplenomegalia. Otros biomarcadores moleculares potenciales incluyen lipoproteínas de baja densidad, osteoactivina, ferritina sérica, progranulina y CCL18. También se han empleado modalidades de imagen como la resonancia magnética, la absorciometría de rayos X de energía dual, la espectroscopia infrarroja por transformada de Fourier y la elastografía transitoria y de ondas transitorias para estudiar las características clave de la EG. Sin embargo, se necesitan más estudios para establecer la eficacia de estas metodologías de imagen. Hasta la fecha, no existe un biomarcador estándar de oro que pueda predecir con seguridad las características clave de la EG. Si bien se han identificado muchos biomarcadores potenciales, cada uno tiene limitaciones inherentes; por lo tanto, los parámetros clínicos continúan definiendo los objetivos del tratamiento y evaluando los resultados. El paciente suele ser su propio control y los niveles a lo largo del tiempo ayudan a monitorizar su tratamiento y normalmente se normalizan con un tratamiento adecuado.
Referencias
1. Baris, Hagit N., Ian J. Cohen, and Pramod K. Mistry. “Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history.” Pediatric endocrinology reviews: PER 12.0 1 (2014): 72.
2. Gary, Sam E., et al. “Recent advances in the diagnosis and management of Gaucher disease.” Expert review of endocrinology & metabolism 13.2 (2018): 107-118.
3. Mistry, Pramod K., et al. “Consensus Conference: A reappraisal of Gaucher disease-diagnosis and disease management algorithms.” American journal of hematology 86.1 (2011): 110.
4. Aerts, Johannes MFG, et al. “Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease.” Current Opinion in Chemical Biology 53 (2019): 204-215.


Lo sentimos, el formulario de comentarios está cerrado en este momento.